
di Alessia Autieri
Partiamo dal sangue
L’eme è un complesso chimico contenente un atomo di ferro in grado di legare ossigeno e fa parte di una famiglia di composti molto importanti chiamati porfirine. Costituisce la parte non proteica di proteine come l’emoglobina, la mioglobina e i citocromi.
È una delle molecole prodotte in maggiori quantità nell’organismo e per questo le patologie che riguardano la sintesi dell’eme possono essere piuttosto gravi.
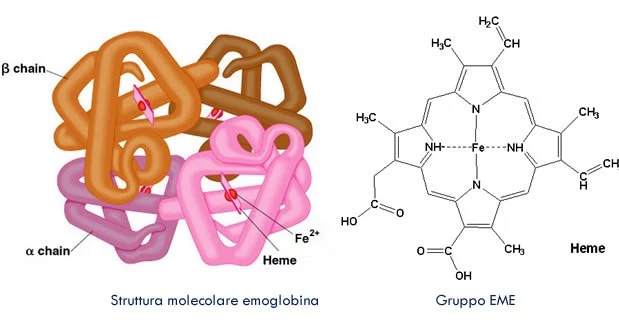
Immagine della molecola di Emoglobina e della struttura del gruppo Eme
Malattie genetiche rare
Le porfirie sono un gruppo di malattie rare, per la maggior parte ereditarie, dovute a deficit dell’attività di uno degli enzimi coinvolti nella via di biosintesi dell’eme. La conseguenza è un accumulo di molecole, chiamate porfirine, o dei loro precursori; queste sostanze, come tali, non sono dannose ma sono fotosensibili. Infatti diventano fortemente tossiche a livello della cute, dove vengono a contatto con i raggi ultravioletti del sole.
Le porfirie vengono classificate in due gruppi principali: acute e non acute. Le forme acute (porfiria acuta intermittente, coproporfiria ereditaria, porfiria variegata) sono caratterizzate da coinvolgimento neurologico. Le forme non acute (protoporfiria eritropoietica, porfiria eritropoietica congenita, porfiria cutanea tarda e porfiria epatoeritropoietica) sono invece caratterizzate da sintomi esclusivamente cutanei tra cui fotofobia.
La pelle esposta al sole presenta scottature, bolle, lacerazioni, croste e cicatrici molto lente nel guarire; può esserci anche senescenza cutanea precoce o alopecia cicatriziale.

Si ha tendenza alla anemia, con emoglobina bassa e ridotto numero di globuli rossi; questo si associa a senso di stanchezza, scarsa resistenza fisica e pallore cutaneo generalizzato. I denti accumulano le porfirine, diventano rossi in trasparenza ed appaiono molto più lunghi a causa del ritiro delle gengive.
Anche a livello oculare i danni possono essere importanti: le ciglia cadono, si ha tendenza alla congiuntivite ed alla sclerite, gli occhi sono cerchiati di rosso per fragilità dei capillari. Spesso è presente splenomegalia: la milza si ingrossa perché il carico di lavoro è molto aumentato per rimuovere i globuli rossi deteriorati dall’eccesso di porfirine. Può manifestarsi anche rachitismo che rende i malati deformi. Inoltre i soggetti porfirinici non possono assumere aglio perché contiene un alcaloide metabolizzato dal citocromo P450 (enzima con un gruppo eme sintetizzato dagli epatociti) che viene così sequestrato; l’organismo reagisce producendo altro citocromo P450 e quindi maggiori quantità di eme, ma il deficit nella sua biosintesi provoca l’accumulo di porfirine e la malattia si acuisce.
Nel passato, tali sintomi hanno contribuito a creare inquietanti misteri intorno alle persone affette da porfiria, tanto più che per curare il pallore causato dall’anemia pare venisse prescritto loro di bere sangue bovino. È sostenuta da alcuni la tesi che sia stata proprio la porfiria, in un’epoca in cui di questa malattia non si sapeva nulla, ad ispirare miti e leggende sulla figura del vampiro, divenuti poi opere letterarie e cinematografiche.
La Protoporfiria eritropoietica (EPP) è la forma più grave di porfiria, le conseguenze di questa malattia possono essere così severe da portare alla morte. Fino a pochi anni fa, l’unica terapia consisteva nella protezione dal sole attraverso creme schermanti e β-carotene. Nel 2008 però è iniziata anche in Italia la sperimentazione di fase III di una molecola in grado di ridurre e, in alcuni casi, eliminare la sintomatologia cutanea della malattia.
Scenesse è il nome dell’impianto inserito nel tessuto sottocutaneo del paziente ogni due mesi, prima e durante i periodi di un’aumentata esposizione alla luce solare. Il principio attivo di Scenesse (afamelanotide) è simile a un ormone presente nell’organismo che stimola la produzione di un pigmento marrone-nero (eumelanina) nella pelle. Questo pigmento è prodotto durante l’esposizione alla luce solare per bloccare la penetrazione della luce nelle cellule; in questo modo Scenesse previene le reazioni dolorose da fotosensibilità.
Nel 2014 la Commissione europea ha rilasciato un’autorizzazione all’immissione in commercio per Scenesse, valida in tutta l’Unione europea. Poiché il numero di pazienti affetti da EPP è basso, la malattia è considerata rara e Scenesse è stato classificato come medicinale orfano (medicinale usato nelle malattie rare).
I malati che avevano preso parte alla sperimentazione sono entusiasti del farmaco: poter passeggiare sotto il sole senza ustionarsi ha innalzato notevolmente la loro qualità di vita. Attualmente però potrebbero dover tornare alle cure con integratori e creme schermanti, infatti la ditta australiana Clinuvel, titolare di Scenesse, ha deciso di quadruplicare il prezzo del farmaco: nel 2010 era venduto al SSN per 5.300 Euro, nel 2018 il prezzo arriverà ai 21.000 Euro. L’AIFA non è riuscita a raggiungere un accordo sul prezzo ed il farmaco è stato classificato in fascia di non rimborsabilità da parte del SNN; esso potrà comunque essere acquistato dalle Aziende Sanitarie delle singole Regioni.
Se siete interessati a vedere un caso reale, si segnala che l’immagine al seguente link è forte e se ne sconsiglia la visione alle persone impressionabili: https://goo.gl/DxmVtY
#noidiminerva #porfirie #vampiri #scenesse
Approfondimenti:
Porfirie: https://goo.gl/ciTzRn
Scenesse: https://goo.gl/jNJPZ7
Credits pic:
https://odobiochem.wordpress.com/2016/04/09/j-e-le-porfirine/





Scrivi un commento